
Research highlights
Chiral chemistry: catalysts benefit from polymer support
In industrial chemistry numerous processes hinge on a chemical’s “chirality”- a feature of the molecular structure that has implications on its chemical and optical properties. A great deal of work has gone into finding ways of selectively producing chiral chemicals. While chiral catalysts can improve production efficiency under milder conditions, so far approaches for retrieving the catalysts from the product have mostly reduced the catalytic activity. Now Shinichi Itsuno, Yosuke Hashimoto and Naoki Haraguchi report how to immobilise industrially important chiral complexes on polymer supports for re-use in effective chiral catalysis.
Chirality describes a type of asymmetry whereby the molecular structure cannot be superposed on its mirror image (like your left and right hands). An example is the sweetener aspartame whose mirror image or alternative ‘enantiomer’ is tasteless. Chiral amines (organic chemicals with an –NH2 group) and their derivatives are particularly important intermediates and building blocks for the synthesis of biologically active molecules.
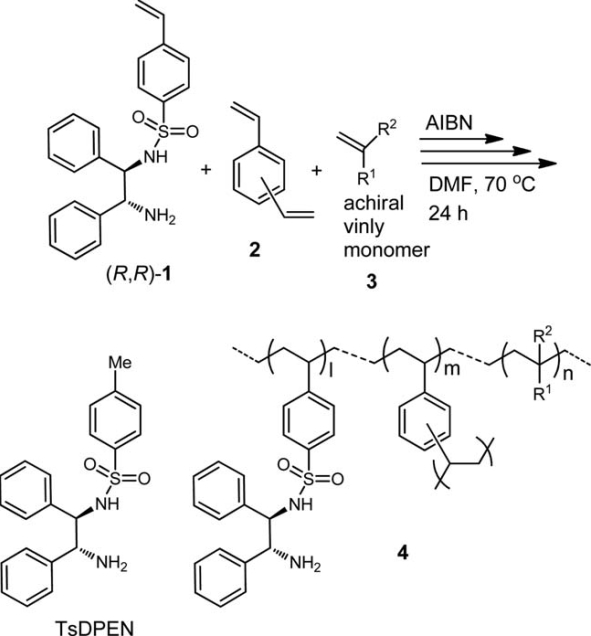
Itsuno and colleagues at Toyohashi University of Technology devised an approach for synthesising chiral complexes of iridium immobilized on a crosslinked polymer to catalyse hydrogenation of cyclic chemicals into chiral amines. Contrary to previous reports where polymer supports have inhibited catalytic activity they found the reaction rates were similar to the non-polymerised complex and the enantiomer selectivity was greater.
Experiments also demonstrated potential for chiral catalytic activity in aqueous environments. As the researchers point out in their report, “To our knowledge, this is the first example of a polymeric iridium catalyst for chiral amine synthesis.”
- Reference:
- S. Itsuno, Y. Hashimoto, and N. Haraguchi.
- Synthesis of chiral iridium complexes immobilized on amphiphilic polymers and their application to asymmetric catalysis.
- Journal of Polymer Science A 52, 3037–3044 (2014)
- doi: 10.1002/pola.27351
Shinichi Itsuno
Enlarge Image
Fig1:Itsuno, Hashimoto and Haraguchi have synthesised an iridium complex crosslinked on a polymer support that can be effectively used to catalyse hydrogenation of cyclic nitrogen-containing molecules into chiral amines. N-toluenesulfonyl-1,2-diphenyl ethylenediamine (TsDPEN)-Ir-Cp* shown on the left still had high catalytic activity when crosslinked with polymers. Polymer crosslinking was achieved by introducing the achiral vinyl monomer ligand (3) in the presence of the crosslinking agent divinyl benzene (2) to produce polymeric chiral ligand (4).